รายการของคุณว่างเปล่า
เพิ่มสินค้าเพื่อขอใบเสนอราคา

The fragility of RNA is well known to anyone who has been in a molecular biology lab. RNA preparations are careful, and perhaps the biggest challenge any researcher faces is contamination by genomic DNA. Genomic DNA (gDNA), which is extracted simultaneously with RNA due to their similar physical and chemical properties, silently contaminates your experiments, generating false-positive results in RT-PCR, causing errors in gene expression quantitation, and rendering all quantitative information invalid.
DNase I is the enzyme that deals with this problem. DNase I is a monomeric glycoprotein that hydrolyzes both single- and double-stranded DNA molecules by cutting them into dinucleotides and oligonucleotides. In essence, it eliminates DNA from your experiments without damaging your RNA preparations. By using RNase-free DNase I, you get two things: DNA elimination with no RNA damage.
Here is how DNase I works, why it matters, how to run the protocol properly, how to handle enzyme inactivation before cDNA synthesis, and what to watch out for at every stage.
Deoxyribonuclease I, or DNase I, is an endonuclease, specifically a non-specific one. DNase I cleaves DNA nonspecifically, generating 5'-phosphorylated di-, tri-, and oligonucleotides. The enzyme can be found naturally in different tissues and body fluids. In the laboratory, researchers usually isolate the enzyme from bovine pancreas. An example of such an enzyme would be DNase I (RNase-free) EN-173S made by XL Biotec – an enzyme isolated from bovine pancreas, available in units of 2000, with a storage temperature of -20°C.
The enzyme is useful in RNA research because it effectively degrades DNA without affecting RNA. DNase I requires divalent cations, especially magnesium and calcium, to function properly. In standard reaction conditions, DNase I acts effectively on single- and double-stranded DNA. That matters because DNA in biological samples exists in different forms.
Besides, the RNase-free DNase protocol of this product is non-negotiable. Traditional DNase I may contain small amounts of RNase, leading to the degradation of the RNA sample that is supposed to be protected by DNase I.

Genomic DNA contamination is not uncommon. Rather, it is an intrinsic issue that exists in almost all protocols for isolating RNA. When scientists use monophasic lysing solutions or column purification methods to extract RNA, traces of gDNA are always present in the extracted RNA, regardless of the protocol used. The scientific literature indicates that RNA obtained from specific organs, such as the spleen, kidney, or thymus, exhibits higher DNA content. In one case, RNA extracted from tumors using the Chomczynski-Sacchi procedure contained approximately 7% DNA contamination by weight.
This problem arises directly in RT-PCR, since PCR technology cannot distinguish between the cDNA generated from your RNA sample and the foreign DNA with a similar sequence. The result is an overestimation of transcript abundance and potential false positive signals.
Numerous studies published in scientific journals have established that a difference of at least 5 quantification cycles between the RT+ and RT- reactions indicates that less than 3% of the total signal arises from gDNA. If this number falls below the set limit, DNase treatment for RNA purification is required. No RNA extraction protocol consistently yields gDNA-free RNA without an active DNase treatment step.
Conducting DNase treatment effectively to purify RNA requires adherence to the proper procedure. The objective is to complete DNA digestion without compromising RNA integrity. Here is how most molecular biology laboratories run it:
One crucial technical point is the inactivation temperature of heat. According to the literature, 5 minutes of inactivation at 75°C does not affect most mRNA, whereas 5 minutes at 95°C deactivates around 80% of the mRNA. Always set the inactivation temperature to 75°C.

Inactivation of DNase I before cDNA synthesis is the most important step in the process, yet it is the one most scientists ignore. Any residual active DNase I in your sample will degrade the newly synthesized single-strand cDNA, thereby spoiling everything. Therefore, inactivating your sample becomes mandatory.
In essence, there are two ways to inactivate DNase I: first, by heat treatment. Place your sample in a water bath at 75°C for five minutes. This temperature is enough to inactivate DNase I, but not your mRNA. You should be careful not to exceed this temperature, because studies indicate that heating your sample to 95°C will kill 80% of your mRNA and the enzyme, rendering all your work worthless.
The second method uses EDTA to chelate metal ions essential for DNase I activity. Add EDTA to your reaction to bring its final concentration to 2-5 mM. This will denature your enzyme without any heat involvement whatsoever. However, EDTA should not be neglected in your subsequent experiments, as it may also affect your RT. S
In certain commercial reagent kits, a unique DNase elimination reagent physically removes the DNase enzyme from the solution without heating or chelation. Such kits prove quite valuable for minimizing sample loss, as even a small amount of RNA can be lost. Whatever technique you decide to use, make sure you run a test on a No RT in your RT-PCR experiment after DNase treatment.
DNase treatment before cDNA synthesis directly determines the quality of your results, and you cannot overstate how much it matters. The RNA sample should be absolutely devoid of genomic DNA before you initiate reverse transcription. This is because the genomic DNA will function as a template during the reverse transcription process, and your final cDNA product will be a mixture of cDNA sequences generated from both your RNA sample and the genomic DNA contamination.
This becomes particularly crucial when studying genes that lack identifiable exon-intron boundaries, such as long non-coding RNAs. For example, a study on one of the most well-researched lncRNAs in cancer research, MALAT1, showed that because MALAT1 lacks exon-intron boundaries, primers designed to detect MALAT1 would also amplify genomic DNA.
In the absence of a stringent DNase I enzyme for RT-PCR preparation, research using such markers might yield false positives that lack biological significance. The same applies to pseudogenes, which are mRNAs that have been reverse-transcribed and integrated into the genome. These molecules lack introns; thus, span primers will not provide any defense.

DNase can be used in two primary methods: either directly on the column membrane during RNA extraction or in solution after the DNase I RNA isolation workflow. Each method has its own strengths and weaknesses depending on the sample, time, and purpose.
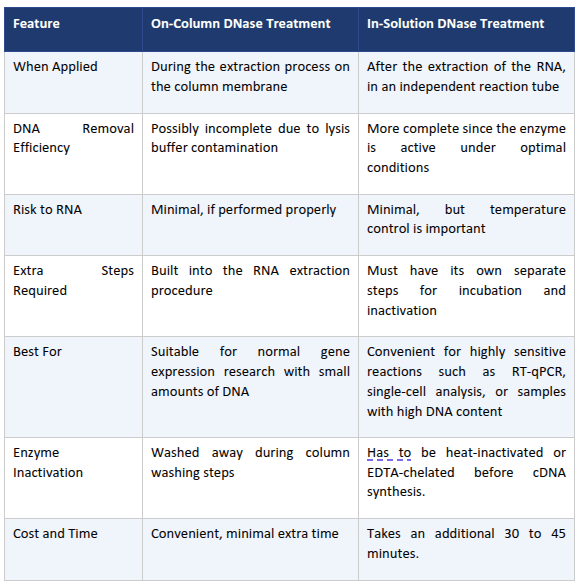
In-solution DNase I treatment provides a superior approach to the on-column DNase I technique. For accurate, quantifiable RT-qPCR or any sensitive assay, treat your RNA in solution with a certified RNase-free DNase I.
Although removal of genomic DNA contaminants from RNA samples is probably the most common application for which DNase I is used, the enzyme serves a broad array of roles in molecular biology protocols. Knowledge of the diverse applications of DNase I will allow you to identify situations where the enzyme might help solve a particular experimental problem that you have not yet thought about being solved through the use of DNase I.
One such use of the enzyme is RNA purification following in vitro transcription. During transcription of RNA using RNA polymerases such as T7, T3, and SP6, the DNA template should ideally be degraded post-transcriptionally to yield RNA transcripts free of DNA contamination. DNase I is usually used to degrade DNA, and this is possible because the enzyme does not degrade RNA transcripts themselves.
Another interesting use case for DNase I is RNA sequencing library preparation following rRNA depletion. Following hybridization of DNA probes complementary to rRNAs and their removal with RNase H, DNase I is used to degrade the DNA probes, thereby providing an rRNA-depleted RNA population for further library preparation.
Also, DNase I can be used for DNase I footprinting experiments, which identify the precise sequences that bind to the protein of interest by partial digestion of DNA-protein complexes. In laboratory studies using cell cultures, DNase I helps decrease the viscosity of cellular extracts by degrading bulky DNA molecules in chromosomes. Lastly, random DNA fragmentation with DNase I is an old but effective approach for generating overlaps in DNA cloning and in vitro recombination.
Not all DNase I preparations are the same, and choosing the correct product for your experiment will give you consistent results compared to those who choose the wrong one. When purchasing DNase I for RNA extraction, there is only one critical characteristic you must look for in the product specifications: DNase I RNase-free enzyme. Regular DNase I preparations contain small amounts of RNases that degrade the RNA being treated, especially during long incubation times.
When considering purchasing DNase I, pay attention to its unit specification, as it varies by manufacturer. Also, confirm the conditions required for enzyme storage and inactivation. If you are using a high-throughput RNA preparation system, ensure the DNase I used is compatible with your equipment.
DNase I serves as the foundation for accurate RNA studies. Your experiments will be compromised without it. Employing a high-quality RNase-free DNase I enzyme, following proper digestion and inactivation procedures, and always performing a no-reverse transcription control should become your three essential practices.
From RT-PCR to RNA sequencing and any lncRNA experiment, DNase I is an indispensable step you cannot afford to miss. Armed with the knowledge shared herein and XL Biotec's DNase I (RNase-free) EN-173S, you can accomplish anything.
1. What is the difference between regular DNase I and RNase-free DNase I?
Regular DNase I can contain trace RNase contamination that degrades your RNA. RNase-free DNase I is certified to have zero RNase activity, making it the only safe choice for RNA work.
2. How do I know if my DNase treatment removed all the genomic DNA?
Run a no-RT control in your RT-PCR. No signal in the minus-RT lane means your DNA removal was successful.
3. Why mustDNase I be inactivated before cDNA synthesis?
Active DNase I will degrade your newly synthesized cDNA. Heat to 75°C for 5 minutes, or add EDTA before starting reverse transcription.
